Phenylketonuria (PKU)
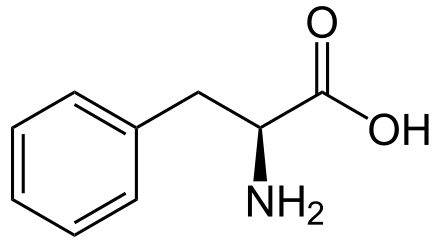
Phenylketonuria (PKU), also known as phenylalanine hydroxylase deficiency or Følling disease, is an autosomal recessive genetic disorder that affects the metabolism of the amino acid phenylalanine. Mutations in the PAH gene result in inefficient or nonfunctional phenylalanine hydroxylase, leading to phenylalanine accumulation, which can be toxic to the brain.
Signs and Symptoms
Untreated PKU can cause a range of severe symptoms, including intellectual disability, seizures, behavioural problems, and mental disorders. Additionally, individuals may exhibit a musty odour, lighter skin, and in infants, microcephaly (abnormally small head). Newborns born to mothers with poorly managed PKU are at risk of heart problems, low birth weight, and other developmental issues.
Genetics
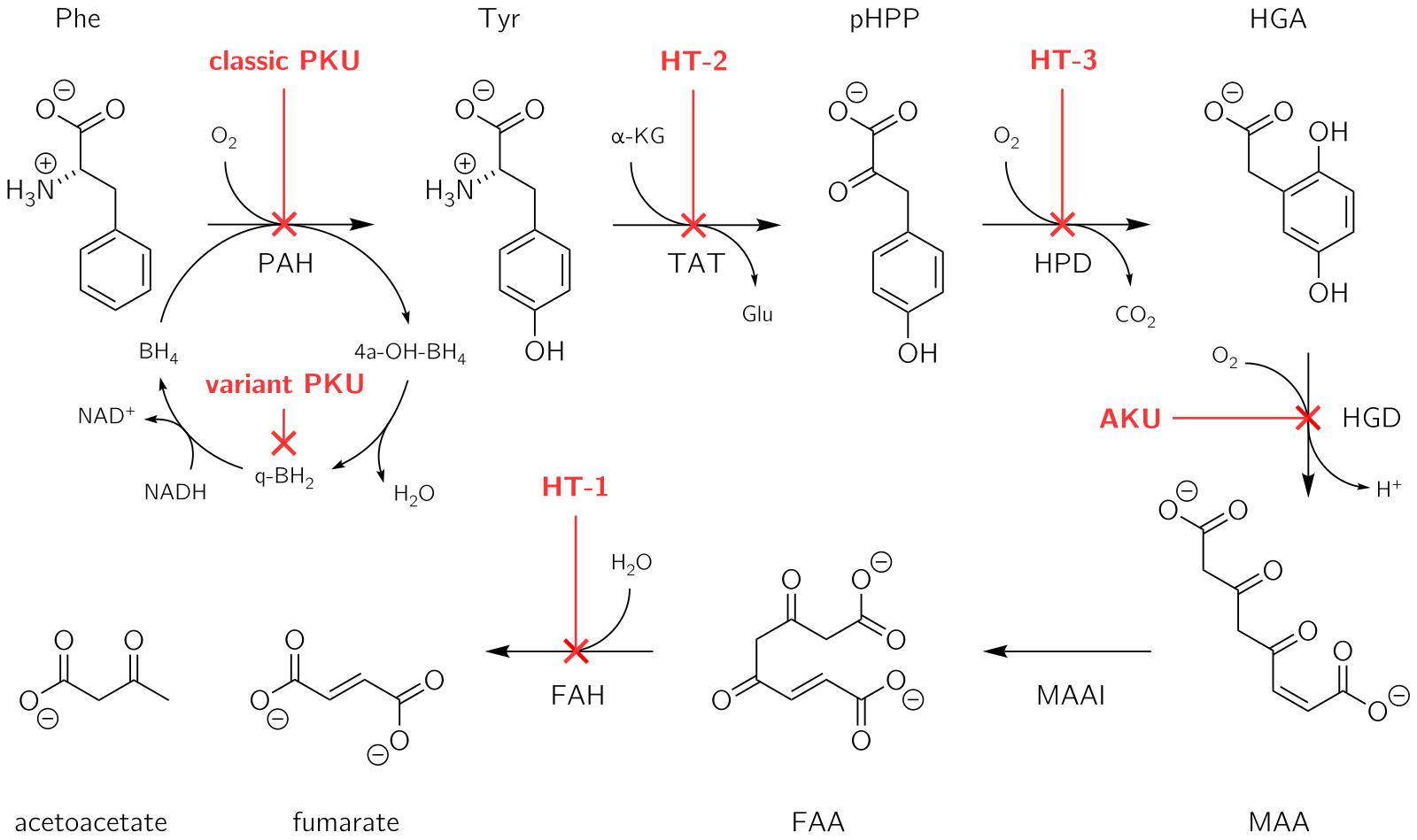
PKU is inherited in an autosomal recessive fashion, requiring two mutated alleles for the disease to manifest. The PAH gene is located on chromosome 12, and over 400 disease-causing mutations have been identified. The enzyme phenylalanine hydroxylase converts phenylalanine into tyrosine, and its deficiency leads to phenylalanine accumulation and subsequent conversion to phenylketones, detectable in the urine.

Pathophysiology
The inability to metabolise phenylalanine leads to its toxic accumulation, causing various neurological and physical symptoms. Classic PKU and its milder forms result from PAH mutations, leading to excess phenylalanine and reduced tyrosine. The high phenylalanine levels interfere with brain development, leading to intellectual disability and other symptoms.
Screening
Screening for PKU is included in newborn screening panels in many countries. A blood sample, usually taken via heel prick 2–7 days after birth, is tested using techniques like the Guthrie test or tandem mass spectrometry to detect elevated phenylalanine levels.

Treatment
Diet
Dietary management is very important in PKU treatment. A low-phenylalanine diet, avoiding foods like soybeans, egg whites, fish, nuts, and certain dairy products, is essential. Fruits and vegetables low in phenylalanine can be consumed in larger quantities. Aspartame, a sweetener containing phenylalanine, must be avoided. Tyrosine supplementation is also necessary due to its deficiency in PKU patients.

Nutritional Supplements
Protein substitute formulas provide essential amino acids and nutrients lacking in a low-phenylalanine diet. They help reduce phenylalanine levels by preventing protein catabolism. Large neutral amino acids (LNAAs) and casein glycomacropeptide (CGMP) are also used to manage phenylalanine levels and improve diet compliance.
Enzyme Substitutes
In 2018, the FDA approved pegvaliase, an enzyme substitute that metabolises phenylalanine, for adults poorly managed by other treatments. Tetrahydrobiopterin (BH4), a cofactor for phenylalanine oxidation, can reduce blood phenylalanine levels in some patients.
Maternal PKU Management
Women with PKU must maintain low phenylalanine levels before and during pregnancy to prevent developmental issues in the foetus. Regular blood tests and strict dietary adherence are essential. As the foetus develops, the mother's phenylalanine intake may need to increase to maintain safe levels.
Epidemiology
PKU incidence varies globally, affecting approximately 1 in 12,000 newborns in the UK. Rates are highest in Turkey (1 in 2,600) and lowest in Finland and Japan.
History
PKU was first described by Norwegian physician Ivar Asbjørn Følling in 1934. The dietary treatment for PKU was developed in the 1950s by Horst Bickel and colleagues. Newborn screening for PKU, introduced by Robert Guthrie in the 1960s, has significantly improved early diagnosis and treatment.
Self-assessment MCQs (single best answer)
What causes Phenylketonuria (PKU)?

Which of the following is NOT a symptom of untreated PKU?
PKU is inherited in which pattern?
On which chromosome is the PAH gene located?
Which enzyme deficiency leads to PKU?
What is the primary dietary restriction for individuals with PKU?
Which of the following sweeteners must be avoided by individuals with PKU?
What is the purpose of the Guthrie test?
Which treatment was approved by the FDA in 2018 for adults with PKU?
Who first described Phenylketonuria (PKU)?
Dentaljuce
Dentaljuce provides Enhanced Continuing Professional Development (CPD) with GDC-approved Certificates for dental professionals worldwide.
Founded in 2009 by the award-winning Masters team from the School of Dentistry at the University of Birmingham, Dentaljuce has established itself as the leading platform for online CPD.
With over 100 high-quality online courses available for a single annual membership fee, Dentaljuce offers comprehensive e-learning designed for busy dental professionals.
The courses cover a complete range of topics, from clinical skills to patient communication, and are suitable for dentists, nurses, hygienists, therapists, students, and practice managers.
Dentaljuce features Dr. Aiden, a dentally trained AI-powered personal tutor available 24/7 to assist with queries and provide guidance through complex topics, enhancing the learning experience.
Check out our range of courses, or sign up now!